
Research Content
Research Overview
When small molecules, particularly ions, transport through complexly intertwined structures, what paths do they take and how fast can they move (conduct or diffuse)? Ion transport phenomena occurring in the nanoscale world play a crucial role in a wide range of fields, from biomolecules in life sciences to polymer materials in materials science.
We use theoretical approaches (e.g., molecular simulations) to elucidate these phenomena, which are difficult to observe in experiments, on challenging spatiotemporal scales. Our goal is to perform theoretical high-efficiency and high-functionality molecular design, and, in collaboration with experimental groups, we aim for broad applications ranging from engineering applications, such as constructing artificial cells and molecular systems, to medical applications, including drug discovery.


Molecular Simulations
Our research is based on methods from theoretical and computational chemistry, particularly Molecular Dynamics (MD) simulations.
To analyze the dynamic properties (dynamics) occurring in biomolecular and polymer systems across a wide range of spatial and temporal scales, it is crucial to use multiscale simulation techniques that combine computational methods from different spatial and temporal scales.
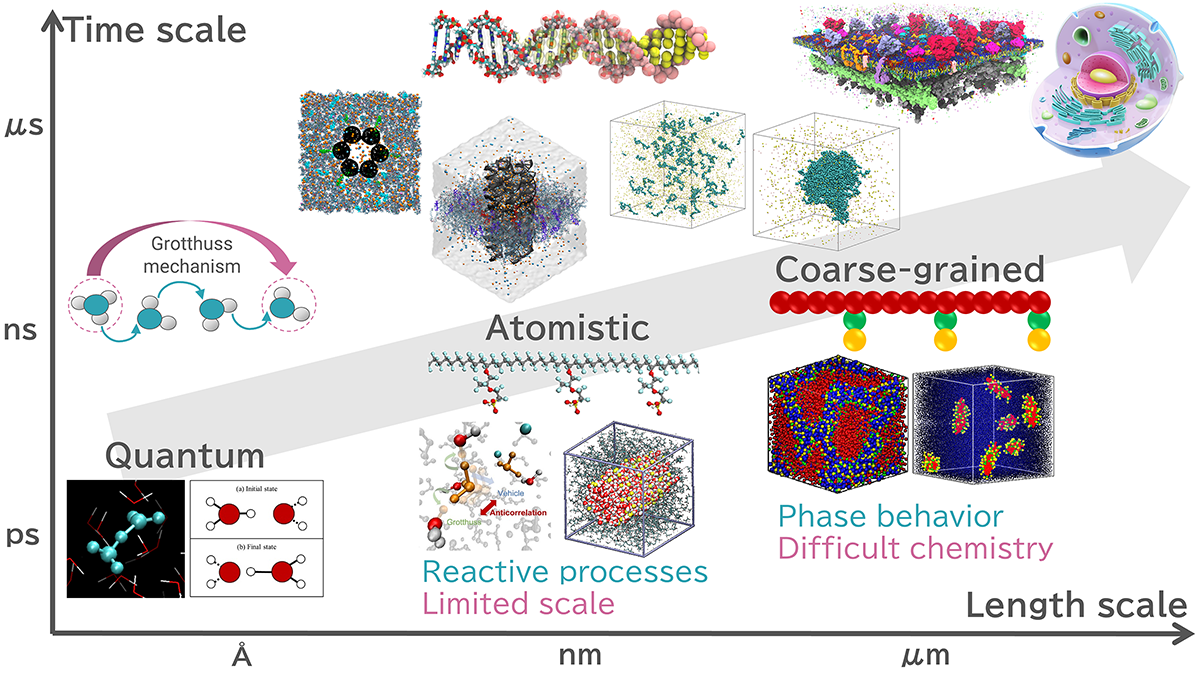
Conceptual diagram of multiscale simulations
-
Quantum Chemistry Calculations
⇒Calculate the electronic states according to the Schrödinger equation of quantum mechanics. -
Reactive MD simulations
⇒Model chemical reactions based on the results of quantum chemistry calculations and handle chemical reactions within the framework of classical mechanics. -
Atomistic MD (Classical MD) simulations
⇒Calculate the movement of atoms according to Newton's laws of classical mechanics. -
Coarse-grained MD simulations
⇒Treat multiple atoms as a single particle to compute phenomena on a larger spatiotemporal scale.